
This meeting was organized by the FDA and the Reagan Udall Foundation at the White Oak Campus in Maryland on October 16th, 2024. The purpose of the meeting was to bring together rare disease patient advocates, academic researchers, biotech industry members, and other key stakeholders to launch the new Rare Disease Innovation Hub. Of over 200 applicants to give public comment, Christine Waggoner, our founder was one of 41 speakers chosen to give a public comment regarding GM1 gangliosidosis.
The topic she was given and the three minute speech she gave are below. In addition, presenters were allowed to show a single slide which is also below.
Full Meeting YouTube Recording
Cure GM1 Presentation Recording
Rare disease-specific (but not application-specific) scientific, regulatory, or policy issues that should be prioritized for consideration by the Rare Disease Innovation Hub
Thank you to the Reagan Udall Foundation and to the FDA for organizing this meeting and creating the Rare Disease Innovation Hub. My name is Christine Waggoner and I am the founder of the Cure GM1 Foundation. GM1 is a neurodegenerative lysosomal storage disease that primarily impacts babies and children.
My daughter Iris suffers from juvenile GM1 and she was diagnosed at age 5 and she is now 16. For 11 years, we have done everything in our power to help the drug development process, raising millions of dollars, publishing papers, participating in natural history studies, conducting a caregiver preferences study, and organizing the first-ever externally-led patient-focused drug development meeting for GM1.
With respect to scientific, regulatory, and policy issues, I would like to highlight three key topics that are relevant to GM1 and other similar conditions.
First, accelerated approval based on primary disease biomarkers, second the ethics of placebo-controlled trials in neurodegenerative diseases, and finally, much needed flexibility for highly heterogeneous populations in ultra-rare diseases.
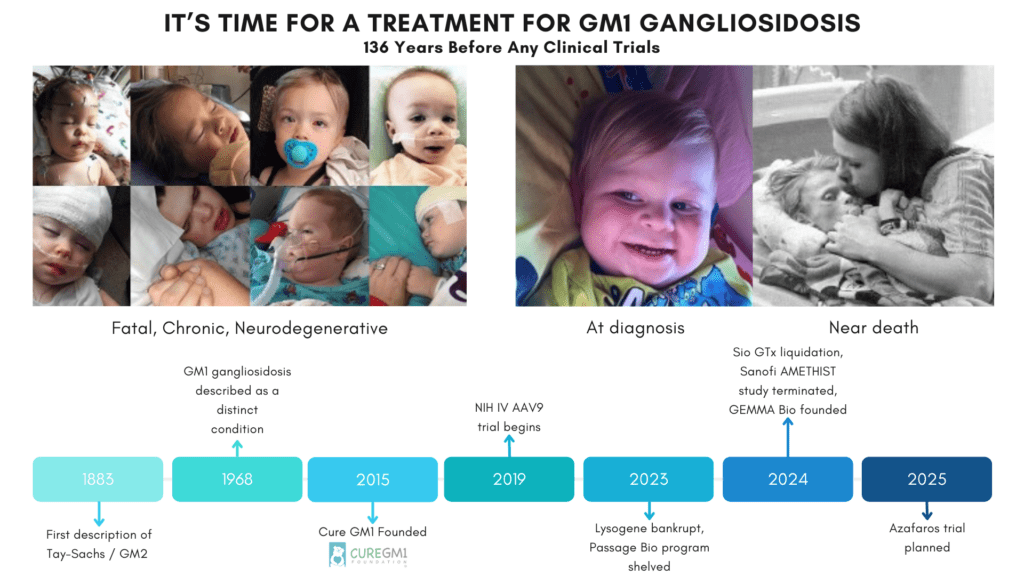
GM1 was clinically distinguished as a unique condition in 1968 and has an incidence of 1 in 100,000-200,000. The deficiency of beta-gal and accumulation of GM1 ganglioside is the very definition of the disease. We must ask ourselves why treatments which have corrected GM1 ganglioside to normal levels are still out of reach of dying patients. Has a framework designed for common diseases caused the abandonment of our programs?
In recent years, 4 companies have gone bankrupt or shelved their programs despite several of the trials demonstrating excellent results in biomarkers and even stabilizing patients and improving quality of life. Accelerated approval based on primary disease biomarkers could save programs that are here now on the precipice of being lost forever.
Children and babies impacted by GM1 are experiencing brain damage, some with degeneration beginning in the womb. Our disease is so severe and the trials are so intense that nearly no family is willing to risk receiving a placebo. The youngest and healthiest children are being asked to give up their fleeting miniscule period of normalcy and to forgo possible treatments that have a reasonable likelihood to help and to stop irreversible brain damage.
Finally, our disease is a spectrum and is extremely heterogeneous and ultra-rare. GM1 requires regulatory flexibility with respect to labeling of products. It is a real possibility that overly strict labeling could prevent swaths of our community from ever having a treatment.
Innovation and flexibility are desperately needed for ultra-rare neurodegenerative diseases like GM1. Let’s create a more compassionate framework of meaningful change based on strong science and be on the right side of history by saving lives. Thank you.